Summary: PCR for Pyromark (50uL reactions for sequencing)
Procedure:
- diluted bisulfite treated DNA to 20ng/uL (or 40ng/uL in the case of sample EV2.28 and the positive control)
- prepped mastermix for the 44098 primers, ran PCR using optimized conditions, performed a gel check - looks good, blank is primer dimer only (resolution on camera is bad)

- prepped mastermix for 733 primers, ran PCR, and stored at 2-8C
Run 5uL of PCR on gel to confirm presence of band at ~300bp
Submit all 18 samples, sequencing primers and sample submission paperwork to Cassie at Fred Hutch
7/24/14
Summary: PCR for Pyromark to determine if primers can amplify unconverted DNA
Procedure:
- ran 2 samples both bisulfite converted and non-bisulfite converted: individuals 28 and 29 (28 had a weak band for primer 44098 and 29 had a strong band)
- ran all 3 primers under their optimized PCR conditions (see previous entry 7/9/14)
- ran them out on EtBr gel

The 733 primers amplify both bisulfite converted and non-bisulfite converted DNA. The 748 primers still not showing much amplification. The 44098 primers can also amplify converted and non-converted DNA. Here though, the amplification is almost not present for either 28 sample, but there is amplification for the 29 sample (both bisulfite and non). I should have run this gel out longer to get the separation between the band and the primer dimer (seen in the blank and oyster 28 lanes)
07/09/14-7/10/14
Summary: PCRs for Pyromark
Procedure:
used previously optimized PCR conditions for primers 733 (56C anneal/3.0mM MgCl2), 748 and 44098 (50C anneal/1.5mM MgCl2)
Mmix prep info here:
http://eagle.fish.washington.edu/bivalvia/070914bisulfite.pdf
Samples were stored at 4C overnight. On 7/10/14, 20uL were loaded into sequencing plate for the Hutch and 5uL were used for running the gel.
Gel Image:
The ladder is a 100bp ladder. The band is very bright for the 733 primers. The band is fainter for the 748 primer and is also subdued by being right underneath the dye front. The 44098 primer gives a good band for sample Ev2.20 and Ev2.29, but the other samples' bands are pretty faint and slightly smaller? than the others.
Gel labeling top of bar: sample number (e.g. 16 = Ev2.16, etc.) The 100% methylated control is labeled with "+" and the blank is labeled "B"

Next steps: Take 20uL of PCR product and 100uL of 10uM sequencing primer to Cassie at the Hutch.
7/2/14
Summary: Repeat PCR from 6/27/14. NOTE: Used different sample only -> Ev2.24 (@130.37ng/ul) prepped at 40ng/uL dilution with 3.06uL of DNA with 6.9uL H20 then did 1:2 serial dilutions to 10ng/uL
Results:
I am happy with the results from the 748 primers. I don't think the starting template amount makes a lot of difference, so will use the 20ng going forward. The 44098 primers are still behaving weirdly. The blank has a faint band that is between 100-200 bp. Since all the reagents are the same in both master mixes, I might assume that the band is from contamination in the primer? Something is definitely amplifying in the DNA wells, but it's smeary. I'll continue to run the gel out and see if it clears up at all. I think I can conclude that the PCR I ran last week did not include polymerase, meaning that the funny band in the 44098 wells was occurring anyway - maybe some kind of primer dimer/timer/... thing?

same gel, run out an hour longer (yes, it was also dropped). Running it out longer helped separate out the primer band seen in the blank with the actual amplified bands. There may be a little bit of smearing above the main band?, but overall it looks good.

Summary/Next steps:
I will order the sequencing primer for all 3 primer pairs and run the PCRs at the optimal conditions for each primer pair. These samples will be sent to the Hutch for pyrosequencing.
6/27/14
Summary: Continued optimization of bisulfite primers (see 6/25/14 for most recent PCR)
Procedure:
- Dilutions of Ev2.28 were performed to load 40, 20, 10 and 5 ng of DNA per reaction (recommendation for bisulfite PCR is 10-20 ng DNA/reaction; previous PCRs used 20ng/reaction)
- prepped master mixes for primers CgBS_748_187112F/R (SRID: 1595/1594) and CgBS_44098_295365F/R (SRID: 1593/1592)
- Mmix prep (1.5mM MgCl2) can be found here: http://eagle.fish.washington.edu/bivalvia/062714bisulfite.pdf
- Anneal temp for this PCR is 50C
NOTE: checked dilutions and realized I only loaded 20, 10, 5 and 2.5ng/reaction for this assay
No bands present in 748 lanes. Low MW band present in all reactions of 44098 - either contam or dimer. One thing I noticed on 7/214 when setting up the PCR is that I almost forgot to add the polymerase because I leave it in the freezer until I need it - maybe I forgot to add? Previous PCR with the 20ng condition produced a band, so something did not go well here. Will repeat and include higher DNA load.

6/25/14
Summary: continued to optimize bisulfite primers (see previous entries in 6/20/14 and 6/18/14 for results)
Procedure:
- prepared master mixes and used a bisulfite treated gDNA sample as template (EE2v2.28 diluted to 20ng/ul) for all reactions
- see details of temp gradients, MgCl2 conc. and play layout here: http://eagle.fish.washington.edu/bivalvia/062514bisulfitepcrlayout.pdf
- ran 20uL of PCR reaction on gel (HyperladderII on top, HyperladderI on bottom)
The band size are as expected 733 =251bp, 748=233bp, 44098=128bp. I am happy with the performance of the 733 primers. All anneal temps look similar so going forward I will use a 56C anneal for these primers. Primer 748 is showing weak bands at both MgCl2 concentrations. In order to ensure the template isn't inhibiting the reaction I will do a dilution of the DNA and run the 50C anneal temp at 1.5mM MgCl2. The 44098 primers are show a very light smear in the region of 128 bp. I will perform a similar dilution series of template at 50C anneal as well to try to improve this.

6/20/14
Summary: a) ran PCRs from 6/19/14 on gel, b) bisulfite treated the EE2v2 samples to be used for the MBD-ChIP validation
a) Gel
Procedure:
- added 6uL of 5x loading buffer to 25uL PCR reaction
- loaded 12uL of each PCR on gel and 5uL of HyperladderII
Unfortunately the resolution on this gel is not as good as the last one. Not sure if it's because I only loaded 12uL instead of 20 or if it has something to do with the gel box or the way I ran it. The band size are as expected 733 =251bp, 748=233bp, 44098=128bp, but I can't tell if there is a single or multiple bands. I will use this gel to limit the temps and MgCl concentrations for one additional optimization assay. For the 733 primers I will repeat the 56, 58, 60 and 62C conditions with the 3.0 MgCl2 conc. For the 748 primers there are no real strong bands, so I'll try lowering the anneal temps. I think I'll try 1.5 and 3.0mM concentrations and do a gradient of 50 - 56C. For the 44098 primers I'll also try a few lower temps and repeat a few temps that showed stronger bands. So with the 1.5mM and 3.0mM concentrations I'll do a 50, 53, 55 and 56C temps.

b) Bisulfite Treatment
Procedure:
followed EpiTect procedure for: Sodium Bisulfite Conversion of Unmethylated Cytosines in DNA (except for sample Ev2.22 where I used the prep described in method for "Sodium Bisulfite Conversion of Unmethylated Cytosines in DNA from Solutions with Low Concentrations of DNA")
Volumes/Concentrations used:

http://eagle.fish.washington.edu/bivalvia/skitch/062014bisulfite.pdf_%281_page%29-20140623-155651.jpg
Results:
Nice 260 peaks. The A260/A280 is really high for these samples. The exact same thing happened when I have done bisulfite treatment in the past. The blank was EB buffer from the EpiTect kit.


6/19/14
Summary: PCR optimization for PyroMark assays (bisulfite). Performed additional anneal temp for primers: CgBS_733_26796F/R (SRID: 1597,1596), and tried out primers CgBS_748_187112F/R (SRID: 1595/1594) and CgBS_44098_295365F/R (SRID: 1593/1592) for the first time.
Procedure:
- bisulfite treated gDNA test sample: a pool of gill and EE2 treated gill (011212) was used for all conditions
- ran PCR on the qPCR machine to get a temp gradient. the following 6 anneal temps were tested: 53, 54.9, 56.1, 59.6, 62.3,64C for the new primers and additional temps:59.6, 62.3, 63.1 and 64 were tested for the CgBS_733 primers
- also ran 3 concentrations of MgCl2: 1.5mM, 3.0mM, 4.5mM for the new primers (only 3.0 for the CgBS_733 primers)
- 1 blank per mastermix at 57.7C for the new primers and 61.1 for the CgBS_733 primers
- used Invitrogen Platinum polymerase, mmix and plate layout here:
6/18/14
Summary: ran the PCRs from 6/17/14 on a gel to look for PCR conditions that give single strong band
Procedure:
- added 6uL of 5x loading buffer to 25uL PCR reaction
- loaded 20uL of each PCR on gel and 5uL of HyperladderII
The band size is as expected @ 251bp. All of the conditions showed amplification with the exception of 1.5mM MgCl2 at 60C. Almost all of the PCRs show a faint band just above the major band. The 'best' condition is 30mM MgCl2 at 60C anneal temp, but I would like to take the anneal temp a little higher and try again. I'd also like to check with Cassie to see if I could gel purify the major band and submit that for sequencing. Next steps is running higher anneal temps with the 3.0mM MgCl2 condition and running the additional 2 primer pairs (also going for a wider range of temps for those as well).

6/17/14
Summary: PCR optimization for PyroMark assays (bisulfite). Starting with just 1 primer pair and running 24 different conditions (anneal temps and MgCl2 conc.'s vary). Primers: CgBS_733_26796F/R (SRID: 1597,1596) note: reverse primer is biotinylated
Procedure:
- bisulfite treated gDNA test sample: sperm P19 bisulfite treated 011212 (diluted with H20 to ~20ng/uL) used for all conditions
- ran PCR on the qPCR machine to get a temp gradient. the following 6 anneal temps were tested: 53,55,56,58.2,59,60C
- also ran 3 concentrations of MgCl2: 1.5mM, 3.0mM, 4.5mM
- 1 blank per mastermix at 57C
- used Invitrogen Platinum polymerase, mmix prep and plate layout here: (link to: http://eagle.fish.washington.edu/bivalvia/gel_and_other_images/06172014qPCRlayout.pdf)
- followed Cassie's suggestion to run 45 cycles (need to use up all of the biotinylated primer, extra will affect pyrosequencing) denature (94C), anneal (variedC), and extention (72C) were all 30s with initial hot start of 10min/95C and final extend 10min/72C
6/16/14
Summary: prepare a 'positive control' DNA that is 100% methylated to use in Pyromark assays
Procedure:
- gDNA used: Ev28 (isolated by Sam) @126.01ng/uL
- prepared duplicate reactions with 0.5ug each following mfr instructions for M.Sss (link to protocol: http://www.zymoresearch.com/downloads/dl/file/id/122/e2010i.pdf)
- incubated at 30C for 3 hrs. then added additional methylase and incubated overnight
- stopped reaction 06/17/14 by incubating at 63C for 20 min
- stored gDNA in Mac's bisulfite DNA box in -20C
3/28/14
Summary: Test gDNA isolation of early gonad sample with Qiagen kit (used day zero female "7.go")
Procedure:
Followed manufacturer's instructions (starting on page 28) of Qiagen DNeasy Protocol (Animal Tisues)
-starting material: 13mg (cut up)
-proK/ATL buffer overnight (4:20pm start, 10am stop - solution was not viscous)
-observed white precipitate when AL buffer was added, loaded all volume onto column
-eluted in 100uL EB 2x (combined elutions for a total volume of 200uL)
Results: There is strong absorbance at A230 (A260/A230 =1). A260/A280=1.86, concentration=93.4ng/uL
Conclusions/Next Steps: not sure if the carryover is ethanol or salt. If ethanol, I need to make sure the column is really dry prior to elution. If salt from the sample, probably not much I can do. I am concerned however about how it will affect the downstream application (bisulfite treatment).
Follow up: Sam ran another test sample (EE2v2.9go) on 4/2/14 and had a better A230. He used ~ 10mg starting tissue. The only differences we noted between procedures is he does all spin steps at max speed and does that final spin step for 10 instead of 3 minutes (protocol says to spin at 20g, but centrifuge only goes to 16g so he upped the time). Perhaps his protocol dries the column better and gets residual ethanol etc. off the column.
2/26/14
Summary: Testing RNA recovery using RNAeasy Plus Mini Kit take 2 (take 1 was done 2/25/14): Sonication and Qiashredder columns.
Procedure:
Following up from yesterday's failed procedure, I tried 3 different things today to optimize the protocol:
1. I could have 'too much' gDNA or RNA in sample, to not overload the columns I used the minimum amount of tissue 10mg and the maximum amount of tissue ~30 mg (27 mg) from a day 0 female.
2. I could have poor homogenization so I tried two different things
a) Qiashredder columns: This is one option for homogenization given in the manual. We have these columns (even though they're old: 2007) so I processed 2 samples (the 10mg and 27 mg samples). I disrupted the samples first with mortar and pestle then ran them through the column (max spin for 2 min). Sample was clear after column.
b) Sonication: Sonication is another option for homogenization given in the manual (even though they say specifically to use Qiagen's TissueRuptor, but we only have a regular old sonicator). I did one sonicated sample with 27mg of starting material (a second day 0 female) And did pulse sonication on ice - about 30, 1 second pulses. The sample was clear after sonication.
3. Perhaps there was not enough enough lysis buffer. Today I used 650ul of buffer (should have used 600 maximum, I didn't read it well enough and used 650uL instead).
Then I followed the protocol the same way I did on 2/25/14 except the RNA column step was performed twice since only 700uL of lysate can be placed on the column at a time.
Results:
Neither the 10mg or 27mg Qiashredder'ed samples showed any RNA recovery (eluted in 30uL H2O)
The sonicated 27mg sample showed some recovery with a very small peak at A260. The sample was reported to contain 11ng/uL RNA (330ng total). This should be enough for an RNA-Seq library even if this quantitation isn't accurate because the concentration is too low for Nanodrop to read accurately. The sequencing lab said they have made successful libraries with 50 -100ng of RNA.
Conclusions/Next Steps
The RNA yield is low compared to what the manual states is possible with this kit (minimum of 4ug of recovery). I am not sure if the issue is with the tissue type? I almost want to try the process one time with a gill tissue sample, where we know we get good RNA recovery, and see what the results look like.
In terms of being ready to run my real samples, I am still a little hesitant. I think I have less than 27 mg for most of these samples - probably closer to 15mg for most samples, which may not result in enough RNA.
2/25/14
Summary: Test RNA recovery in the tiny EE2 day 7 gonad samples I have. Using 1 female samples and trying out the RNeasy Plus Mini Kit
No recovered RNA from this test.
Procedure:
- Started by getting a rough estimate for how much tissue I have for each sample. With the exception of individual 22, which is on the array, I have about 10-15mg of tissue for each sample. This should be sufficient for the kit, which uses between 10-30mg tissue
- The Kit manual says you can get between 4- 60ug of RNA from 10mg of tissue (depending on tissue type). At the very, very minimum I need 50-100 ng of RNA to make a library
- I started with sample 26 (day 7 female that was not on the array) as a test sample to see how much RNA I could recover. I had 15mg of tissue to start
- added 10uL of betamercaptoethanol (BME) to 1mL Buffer RLT Plus (make fresh every time)
- added 44mL EtOH to Buffer RPE per bottle instructions
- added 350uL Buffer RLT Plus (with BME) and 15mg tissue and homogenized (~1min) w/ mortar and pestle. Solution was cloudy no solid tissue remained
- Centrifuged for 3min at max speed and removed lysate -- here I noticed a thin, white film on the top of the solution and a tiny 'pellet' at the bottom. I tried to remove just the clear supe. Not sure if the white film on top indicates it wasn't mixed well enough?
- Removed lysate and added to gDNA Eliminator column, centrifuged max speed (says >8000 x g) for 30 s
- Saved flow through and added 350uL freshly prepared 70% EtOH with water supplied in the kit
- Mixed well by pipettng and transferred entire 700uL to RNAeasy spin column and centrifuged for 15s
- Washed 1x with 700uL W1 (centrifuged max speed (says >8000 x g) for 15 s
- Added 500uL Bufer RPE, spin max 15s
- Added 500uL Buffer RPE, spin max for 2 min
- Put column in new collection tube and did Option step 10 - centrifuge 1min max speed
- Placed column in new collection tube and added 30uL RNAase free H20. Let it sit for 1 min and centrifuged for 1min at max speed (still says greater than or equal to 8000 x g)
- Did not perform step 12 using another 30uL of H20
- Put 2 uL on nanodrop with H20 blank and no peaks were observed (0.7ng/uL RNA)
The troubleshooting section of the manual says a few things could be happening here: 1) not enough disruption - try using the sonicator for 30s, 2) reduce the amount of starting tissue OR add more lysis buffer. Too much RNA can clog the column, I am using 1/2 of the maximum amount of tissue, so I don't really think using less is a good idea. I could increase the lysis buffer volume. The instructions say 350-600uL and I used the minimum. Maybe this would help with that weird thin layer I saw after centrifuging the disrupted tissue and Buffer RLT plus.
7/26/13
Summary: purify gDNA (i.e. improve A260/A230 ratio) using Qiagen MinElute Kit samples for control and EE2 treated pools for use in microarray.
Procedure: starting w/ gDNA from 7/21/13
- following Qiagen MinElute protocol for 'PCR purification' added 60uL (5x vol) PB buffer to the samples from 7/21/13
- followed remaining mfr kit protocol with the exception that I left the PE buffer sit on column for 5 min prior to spinning (this step is in troubleshooting guide to help remove salt)
- eluted in 10uL EB and quant on nanodrop
The A260/A230s are much improved (1.9), the A260/A280s are still good = 1.8 - 1.9
Total DNA for the control sample: 54.04 x 9.1uL recovered = 492ng
Total DNA for the treated sample: 56.34 x 10uL recovered = 563ng


Conclusion/Next steps:
The kit worked to improve the A260/A230 ratio. The total DNA recovered is at the low end of what is acceptable for labeling. I will send Cassie at FH this information and see what she says about doing any QC for these samples.
7/21/13
Summary: complete EtOh precipitation from 7/19/13
Procedure: spin max at RT 15min, wash 2x in cold 70% EtOH (spin 5min and remove wash), resuspend DNA in 12uL EB buffer (Qiagen). Quant on Nanodrop
Results:
.jpg "external image 072213nano.bmp%20(2%20documents,%202%20total%20pages).jpg")
.jpg "external image 072213nanoplot.bmp%20(2%20documents,%202%20total%20pages).jpg")
Conc. Next Steps:
It appears that the DNA was still in one of the wash steps/or binding buffer. Bad news is that the A260/A230 is still low. Ordered MinElute kits to try to purify the sample. Threw away the remaining kit supplies from the Zymo Research sample kit
7/19/13
Summary: attempt to cleanup DNA to reduce A230 reading from control and treated EE2v2 pools - no DNA in eluate, attempt to precipiate DNA from washes/binding buffer
Background: these are the input v input fragmented DNA pools from EE2 treated and control individuals (n=3/pool). This material went into the MBD procedure and the methylated fractions were generated from this material. I am trying to improve the A260A230 ratio prior to labeling for the microarray
Procedure: Used the Zymo Research DNA Clean and Concentrate Kit (we had an unopened sample box in the lab)
- 2x vol of binding buffer: 400uL for EE2 exposed, 112uL for control
- follow rest of mfr protocol (basically spin, remove flow through (I saved), wash 2x and remove flow through (saved)
- elute in 25uL EB (Qiagen)
- blanked w/ EB and spec'd on Nanodrop and there was no DNA in the eluate
- tried heating EB to 65C (per protocol for troubleshooting) and eluting in 10uL - no DNA in eluate
- tried to get a spec reading of the binding buffer or wash flow through, but couldn't get a good blank on these reagents
- combined all eluate, wash, binding buffer flowthrough fractions and performed chloroform extraction
- equal vol chloroform
- vortex 30 sec and spin max 15min
- 2.5vol EtOH and 1/10th vol NaOAc
- mix by inversion then placed in -80C
7/2/13
Summary: initiated DNA isolation for EE2v2 sample #22 (day 7 female, EE2 treated). Need additional DNA to make pool for the 'input v input' array assay.
7/1/13
Summary: pos control PCR from MBD procedure 6/28/13, and EtOH non-captured fraction from the control sample and captured wash fractions from the EE2 treated and control samples
Procedure:
- PCR: pos control PCR for captured and non-captured fractions per mfr. protcol (and see 6/14/13 entry for details)
- EtOH precipitation
- EtOH precipitation of non-captured fraction for the control sample and spec for concentration, also EtOH and spec the captured wash fractions to see if can get any more recovery for the methylated fractions
PCR (gel)
The methylated DNA spiked into the pos control sample was present in the captured ('cap') fraction as expected indicating the methylated DNA has been fractionated out. There is a faint band for the nonmethylated spike in the non-captured fraction. It was expected that this band would be much brighter, however, since the non-methylated DNA is NOT in the captured fraction, it's ok that the the band is faint.

Spec of non-captured and wash fractions:
The wash fractions did not have a peak at A260 therefore the samples will not be pooled with the captured material. Overall, there was very little recovery in the washes, most of the DNA is recovered in the original capture step with the high salt elution.
Total recovery for the MBD control sample:
Input: 16ug
Noncaptured: 68.79ng/uL x 180uL = 12.4ug
Captured: 0.947ug
Total yield: (12.4ug + 0.947ug)/16ug * 100 = 83% recovery ~ this is within the range of previous recoveries, again the % DNA recovered in the methylated fraction is less than what is usually found: 7% here, previously found 18-22% of recovered DNA in the methylated fraction. Perhaps the female gonad samples are less methylated than the gill tissue?

6/29/13
Summary: ethanol precipitation of pos controls and EE2 treatment and control samples (initiated 6/28/13)
Procedure:
- followed precip. procedure in MethylMiner kit
- spec'd on Nanodrop

- there is a total of 60uL/samples
- EE2 treatment sample: 15ng/uL x 60uL = 0.900 ug total
- EE2 control sample: 15.79ng/uL x 60uL = 0.947ug total
Minimally, I needed 1ug to do the labeling for the MBD-Chip. I will have enough to do the labeling, but not enough to QC the sample (size/concentration check) first so it will be kinda risky. One way to do a little QC is to send plenty of DNA for the unenriched 'input v. input' samples, which is the same pooled DNA prior to enrichment.
Next: Run the pos control PCR for the MBD procedure and precipitate the non-captured fraction for at least one of the sample to look at total recovery from the procedure.
6/28/13
Summary: MBD repeat of 6/19/13 - EE2 treatment and control sample
Procedure:
- prepping reactions for 16ug of DNA (hoping to get at least 10% back in methylated fraction)
- Prepped beads per MethylMiner kit instructions: a) 160uL beads for samples, 10uL beads for pos. control b) 112uL MBD for samples, 7uL MBD for pos. control)
- 188uL EE2 pool at 78.4 ng/uL (I thought I had 16ug total, but in fact had less so added 14.7ug of DNA)
- 217 control pool at 73.7ng/uL
- Followed remaining mfr instructions to obtain non-captured (performed 4x washes) and captured (high salt only) fractions.
- Proceded to EtOH for all of the captured and non-captured fractions. Added 3M NaOAc and 100% EtOH per manual.
- took 2 uL of the captured fractions out to spec before EtOH precip. Blanked on the high salt buffer and there was no absorption at A260. If only 10% of starting DNA was in there it would be ~5ng/uL, so maybe it's not surprising in that much volume. After EtOH precip will go from 400uL to 60, so we'll see.
- Samples stored at -80 to finish EtOH precipitation (remaining wash fractions were stored at -20C)
6/26/13
Summary: fragmenting DNA for MBD (processing additional DNA from samples 6/11/13 to supplement the remaining DNA)
Procedure:
- EE2: sample 22 is limiting (only 2.4ug of DNA remaining) so pooling 3 individuals in equal quantities (i.e. 2.4ug each)
- sample 22: 35uL @68.52 ng/uL
- sample 20: 11.6uL @206.26 ng/uL
- sample 16: 31.5uL @76.29 ng/uL
- added 21.9uL H20 for total vol of 100 and placed in covaris tube
- Control: sample 32 is limiting, pooking 3 individual in equal quantities (4.85ug each)
- sample 32: 52uL @ 91.51ng/uL
- sample 28: 49.1uL @98.74ng/uL
- sample 29: 33.4uL @ 145.20ng/uL
- added 64.5uL H20 for total vol of 200 and placed 100uL each in 2 tubes.
- sheared DNA using the same protocol as 6/11/13
- pooled the newly sheared EE2 (100ul) and control (total vol 200uL) DNA into the tubes containing the previously fragmented DNA and spec'd
- EE2: 78.4ng/uL ~188uL = 14.7ug
- control: 73.7ng/uL ~ 310uL = 22.8ug
6/20/13
Summary: complete EtOH precipitation of MBD fractions initiated 6/19/13
Procedure:
- followed mfr instructions for EtOH precipitation for captured and non-captured fractions from yesterday's fractionation
- spec'd on nanodrop
- performed qPCR to check pos and neg controls - see 6/14/13 entry for protocol followed (used diluted control DNA from yesterday for the pos spikes)

- Total Yields: for two of the samples: control A and treatment A I precipitated the unmethylated and methylated fractions to look at total recovery. All samples are solubilized in 60uL DNAse free H20
- control A: 7.8uL nonmeth + 0.04ug meth = 7.84ug/8ug input = 98% recovery (0.5% methylated fraction)
- treatment A: 7.3ug nonmeth + 0.3ug meth = 7.6ug/8ug input = 95% recovery (4% methylated fraction)
- Total DNA for methylated fractions: control: 0.5ug, treated: 0.4ug (these are probably not accurate Nanodrop readings since conc. so low)
- PCR results (nonmethylated primers on left, methylated primers on right)

Conclusions/Next steps:
- Previous MBD fractionations had total yields between 63-130% so this was within the expected range
- However, previous runs also had about 18-22% of the total yield in the methylated fraction
- todays fractionation was about 10x less than previous ~2% methylated.
- It appears that the methylated DNA from the control was lost somewhere. Separation did happen since the methylated DNA was not present in the non-captured fraction, but either through loss of pellet? or overdrying of DNA? the methylated fraction was lost. This could also be the case for the samples, where low recovery is due to issue in EtOH precipitation either through loss of pellet or overdrying.
- This is the 5th time I've performed this procedure and the first time this has happened. Results were as expected with the control when I ran it late last week. I used different stocks of ethanol than I did last week and had more samples to manage so I am thinking something happened during the EtOH precip. I observed pellets in all samples after the first centrifugation step, but after the first wash pellets were really hard to see in the methylated fractions.
6/19/13
Summary: Methylation enrichment of Ev2 samples (pooled EE2 treated and control gonad samples (n=3 each))
Procedure:
- Performing 2 replicate MBDs for each pooled sample with 8ug DNA/replicate (33ug DNA processed total 32 sample +1ug pos control)
- 3 individuals were pooled after fragmentation (see 6/11/13), spec'd samples on nanodrop:
- Control pool: 84.6ng/uL
- EE2 pool: 75.5 ng/uL
- Prepped beads per MethylMiner kit instructions: a) 80uL beads for samples, 10uL beads for pos. control b) 56uL MBD for samples, 7uL MBD for pos. control)
- Added DNA to prepped beads
- 94.6uL each control replicate
- 106uL each EE2 replicate
- Followed remaining mfr instructions to obtain non-captured (performed 4x washes) and captured (high salt only) fractions.
- Proceded to EtOH for all of the non-captured fractions, the non-captured and captured fractions for the pos. control, and 2 replicate each (EE2 and control) of the non-captured fractions to evaluate total yield. Added 3M NaOAc and 100% EtOH per manual
- Samples stored at -80 to finish EtOH precipitation (remaining wash fractions were stored at -20C)
6/14/13
Summary: Testing MBD kit by running the control (day 2 of 2)
Procedure: PCR of control DNA using primers specific to the control methylated and non-methylated DNA spikes
- prepped 10uM stock of primers (3uL 100uM stock, 27uL H20)
- prepped new dilutions of spike methylated and non-methylated DNA (1uL into 99uL of H20)
- prepped mastermixes for methylated and non-methylated primers: 6uL primer, 150uL Apex Mastermix, 138uL H20
- added 49uL of mmix and 1uL of the following for each primer set
- pos: 0.5uL diluted methylated DNA, 0.5uL diluted non-methylated DNA
- blnk: 1uL H20
- NC: 1uL non-captured supernatant (non-methylated DNA)
- Wsh: 1uL pooled washes (non-methylated DNA)
- Cap: 1uL of the captured and eluted DNA (methylated DNA)

Conclusions & Next steps:
The results are as expected. The non-captured and wash fractions amplified with the non-methylated primers and the captured fraction amplified with the methylated primers. The next step is to process my samples early next week. I'll need to check before I start and make sure there is sufficient reagents to process 40ug of DNA.
6/13/13
Summary: Testing MBD kit by running the control (day 1 of 2)
Procedure: followed the MethylKit protocol for running the control sample only (DNA provided from kit with methylated and non-methylated DNA spiked in). Stored captured and non-captured fragments @-20C
Next steps: PCR for the methylated and non-methylated DNA. If all goes well will MBD my samples on Monday.
6/11/13
Summary: pooled gDNA from Ev2 isolations on 6/3/13 and sheared pooled DNA
Procedure:
- working backwards with my math, I will need 3 ug of DNA (1ug total DNA, 2ug methylation enriched (MBD) DNA) for the EE2 treated sample (n=3 pooled gonad) and the control sample (n=3 pooled gonad). To get 2ug of MBD DNA I need to start w/ about ~20ug of total DNA since the methylated fraction is only ~13%. So I prepped a pool of 22ug of treatment DNA and 25ug on control DNA (these were a bit higher in concentration so I prepped some extra to run sheared DNA on gel).
- EE2 pool: 96.1uL (#16), 35.6uL (#20), 107.0uL (#22), I then added 61.3uL of EB buffer for a total vol. of 300uL and pipetted 100uL into each of 3 crimp cap Covaris tubes
- control pool: 84.4uL (#28), 57.4uL (#29), 91.1uL (#32), I then added 67.2uL of EB buffer for a total vol. of 300uL and pipetted 100uL into each of 3 crimp cap Covaris tubes
- Sheared each of the 6 microtubes on the Covaris using Rhonda's protocol for 500bp shearing: Duty Cycle 5%, Intensity 3, Cycles/Bursts: 200, Bath: 5C, first treatment is 60sec, second treatment is 30sec
- Pooled the 3 Covaris tubes for each sample back into a microcentrifuge tube and spec'd on Nanodrop
- EE2: 74.4ng/uL (x300uL = 22.3ug)
- control: 81.3ng/uL (x300uL = 24.4ug)
- Ran 0.5ug of sheared control DNA on a 1% agarose/EtBr gel to check size distribution..

Conclusion: most of the DNA is between 500 - 50bp, but a majority of it is between 200-300bp. I think this is ok because Cassie at the hutch says: 'Yes, 500bp is fine…we just want the majority of your sample to be greater than 200bp.' Next step is MBD, but since Claire is having issues with the control samples, I will run the controls myself first to help her troubleshoot and make sure everything is working ok before I run the protocol with my samples.
6/3/13-6/4/13
Summary: gDNA isolation Ev2 day 7 female gonad samples
Procedure:
- isolated DNA from 3 females from control group (2 tanks total) and 3 females from the treatment group (2 tanks total) using DNAzol
- 0.5mL DNAzol
- used 1/2 of the gonad sample from -80C (~8-15ug)
- little pestle action, 2.35uL proK (18.35ug/uL), another 0.5mL DNAzol and O/N incubation @ RT
- followed mfr protocol for precipitation and washing then solubilized in 150uL EB Buffer (Qiagen) and spec'd (samples in June2012 gDNA box)
- spec results:

- 260/280 looks ok, but the 260/230 is quite low. The quantities are sufficient for MBD (I want to have ~7ug sheared for MBD, and 100uL max volume in Covaris tube, so I'll be ok there). Also will need some left over for the 'input v input' assay. The next step will be shearing the gDNA and also confirming design (i.e. poor before or after MBD) and total quantity needed for labeling. I don't want to use more starting material than necessary since MBD kits are limited.
3/29/13
Summary: Ev2 update - sampling 3/25/13/3/26/13
- sampled all remaining oysters day60/61 of trial: 230 total
- froze (-20C) a 60mL water sample from control tank 2
- all measurements can be found in Dropox>Lab>Mac>EE2 trail>'EE2v2 data'
- will keep track of stats performed in Evernote notebook
3/19/13-3/21/13
Summary: isolation and shipment of DNA samples from F1 of vinclozolin treated and control parents to Nanostring. 8 Samples total.
Procedure:
- isolated DNA from ~20mg of gonad tissue from 8 males (4 with vinclozolin treated parents: #3,4,20,21; 4 w/ control parents #44,45,57,58) using DNAzol following mfr instructions. Solubilized in 200uL of PCR H20.
- Did an EtOH precipitation of all 8 samples (only 1 sample had a low concentration (quant by Nanodrop), but processed all 8)
- Quant. after EtOH precipitation:

- Shipped ~10ug of each sample to Nanostring. Remaining DNA stored in Mac's July gDNA box in -20C.
3/20/13
Summary: Ev2 update
- 3/18/13 sampled 2 control animals (tank 4): 1 undetermined and 1 male by light microscopy
- 3/20/13 total degree days for this trial =1107 -> this is what I was initially targeting as 1100 degree days should be 50% mature ova (Muranaka and Lannan 1984)
- 3/20/13 samples 2 EE2 treated animals (tank 1 and tank3): 2 were female

2/1/13
Summary: sampled 1 week time-point for Ev2
Procedure:
- sampled 15 oysters from 500ng/L EE2 tx and 15 oysters from control tank (n=5/tank)
- samples taken: gonad (underside of animal), histology cross section between muscle and labial palps, and gill sample. NOTE: gonad samples are quite small. These animals were putting on glycogen (white tissue), but no gonad tubules were present. I looked at a few gonad samples under the microscope and no gametes were observed.
- measurements for this time-point were added to the original spreadsheet. See link from 1/25/13
1/31/13
Summary: shipped T=0 histology samples for Ev2 (see 1/25/13)
1/30/13
Summary: completed DNA isolation from 1/29/13 - note: going to repeat samples with less starting tissue, max amount of tissue resulted in super viscous solution that was not acceptable for downstream steps.
1/29/13
Summary: DNA isolation from male gonad tissue from the vinclozolin F1's
Procedure:
- 4 vinclozolin males (3 & 4 (10 x 11 cross), 20 & 21 (10 x 17 cross)
- 4 control males (44 & 45 (19 x 31 cross), 57 & 58 (19 x 30 cross)
- 40-50mg of tissue/1mL DNAzol/400ug proteinaseK (exception: sample 3 only 500uL DNAzol
- rotating O/N @ RT
1/25/13
Summary: initiated 'Ev2' trial, EE2 exposure
Procedure:
- prepped 10mg/mL EE2 stock solution: 0.06g in 6mL EtOH and stir at RT for 1hr
- prepped 0.02 mg/mL working stock: 10uL 10mg/mL EE2, 4990uL DI H20 (prepped control working stock 10uL EtOH, 4990 uL DI)
- treatments: 750uL working stock into 30L of water for a final concentration of 500ng/L EE2
- oysters: diploid oysters from Thorndyke bay (mixed families) spawned in Summer of 2012
- 3 replicate treatment tanks, 3 replicate control tanks: 50 oysters each (150 oysters/treatment). Tanks are 30L seawater, single pump and airstone. Starting temp 11C (recording feeding, temp change and water changes in paper notebook in wet lab)
- sampled 10 oysters at T=0 (gill, gonad, histology), size here
12/19/12
Summary: Sampled F1 generation of vinclozolin exposed oysters and control oysters
Procedure
- 2 vinclozolin treated families (male (#10) mated with 2 females(#11 & 17)) crosses: 10x11 and 10x17
- 2 control families (male (19) mated with 2 females (#30 & 31) croses 19x30 and 19x31
- sampled 15 animals from each family measured: length, width, total weight, sex determination and sampled gill and gonad (sampled kept on dry ice and stored on top shelf of the -80)
- morphometrics here: https://docs.google.com/spreadsheet/ccc?key=0AuvyLilchDgFdEozd3Z0TTVDUWtwajUwUUhvcjBBT1E
- also measured all oysters from each family on 09/26/12. Data can be found on same worksheet but "09/26/12" tab
- NOTE: on 11/16/13 most of the remaining oysters were dead (19x31 one oyster remaining, 10x17 2 oysters, 10x11 7oysters, 19 x 30 10 oysters)
10/23/12
Summary: took a few oyster samples during FISH441 lab today to have in the freezer. C.gigas gonad samples (male and female) and O.lurida samples gill and gonad. The Olys had been exposed to an acute heat shock 40C for 1hr. When sex could be determined it was written on the top of the tube. These samples are stored in the top shelf of the -80 in 'Mac's -80C' box.
9/5/12
Summary: run remaining PCR product from samples analyzed 8/29/12 on gel

same gel closer up:


8/29/12
Summary: ABI3730 run at Manchester
Procedure: 1uL of PCR product was loaded into wells containing mastermix of LIZ500 size standard and Hi-Di formamide.
Plate layout: row A: primer set 1, row B: primer set 2 etc.. through G, row H: no template; column 1: Blanks, column 3: 152M, column 5: 152H, column 7: 91M, column9: 91H, column 11: no template
8/28/12
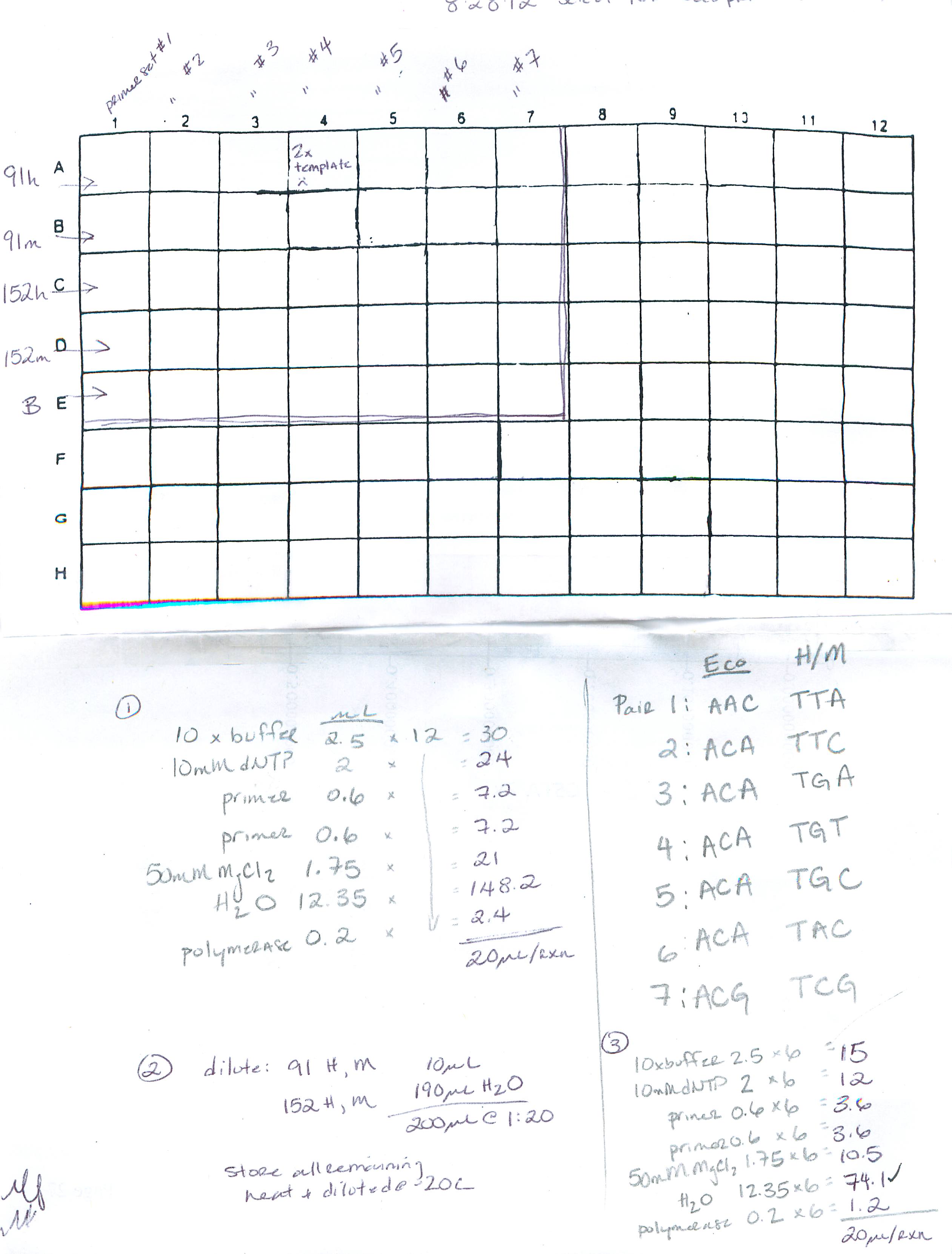
Summary: perform pre-select and select PCR (using FAM labeled primers) for test run of MSAP on ABI sequencer
Procedure: 1) perform pre-select PCR (2 samples: 91 (control) and 152 (EE2 tx)), cycling params: 'PRESELECT' 2) dilute pre-select product, 3) select PCR using FAM labeled Eco primers, cycling parameters: 'TD/56C'. See deets here:
http://eagle.fish.washington.edu/trilobite/Sites_genefish_100112/Mac/082812%20procedure_01.jpg
{kind=link}
8/25/12
Summary: MSAP select PCR - testing amplification after pre-select PCR using Amplitaq
Procedure:
- samples: 91H pre-select product neat, 91M pre-select product neat, 91H and M pre-select PCR products at a 1:20 dilution
- a positive control was also prepared using pre-select product from 7/27/12 (Apex mastermix)
- negative controls included 5uL of template of the blank used in the pre-select PCR and a 5uL of H20

The pre-select PCR using AmpliTaq polymerase does amplify bands even though a smear was not observed on a gel run previously (8/7/12). If you compare the banding pattern between the + control (Apex) and the Amplitaq (can only do this for the HpaII ("H") samples), it appears that most of the bands are similar, but they are of different intensities and some bands are observable using the Amplitaq that were not observed using the Apex. Because the AmpliTaq is a higher quality polymerase, I think going forward will give more reproducible results than with the Apex. There is a single band that amplified when the blank from the pre-select PCR was used as template. Oddly, the same size band was not observed in any of the samples. Possibly there was contamination after the PCR was performed and prior to running the select PCR? The select PCR blank was clean. Of note, the Hpa bands are more clear than the Msp bands for this particular primer set. The Msp lanes still show some high background.
8/21/12
Summary: MSAP pre-select PCR with Amplitaq (3.5mM MgCl)
Procedure: performed pre-select PCR using sample 91 (both H and M dig-lig). Previously (8/7/12), the pre-select PCR using Amplitaq did not show up as a smear on the gel. In order to see if amplification had occurred I needed to repeat the pre-select here and then add this product as template to the select PCR. An aliquot of PCR product was used to prepare a 1:20 dilution (in H20) and all samples were frozen at -20C.
8/7/12
Summary: run PCR products from 8/6/12 (see 'Next steps') on gel: MgCl2 titration using Amplitaq polymerase.
Results: This images shows the results of the Metaphor agarose gel (~2hr at 100V). First, I am having a really difficult time getting the comb out of the gel without breaking some of the wells (as evidenced by the overflow). Gel issues aside, the blanks are clean and amplification was observed using 3.5 mM and 4.5 mM MgCl2, but not 2.5mM MgCl2. The 3.5 mM MgCl2 lane shows the best resolution of bands and should be used going forward. The pre-select PCR's did not show an observable smear (not included in image below, loaded to right but all lanes are blank). Since I added loading dye to the entire pre-select PCR I will have to re-run to check and see if enough amplification occurred to get bands on the 2nd round of PCR. If I am able to do a test run tomorrow on the sequencer I will use the pre-select products generated 7/27/12 as template in the select PCR.

8/6/12
Summary: results of Amplitaq PCR from 8/3/12, troubleshooting Amplitaq results
Procedure:
- On 8/3/12 a test run of select (primer set 3) and pre-select PCRs were performed using Amplitaq polymerase (1.5mM MgCl2) these were run on a gel and no amplification was observed (Apex was also tested (4.5 mM MgCl2) and produced the expected banding pattern)).
- After comparing the reaction components, I noticed that the salt concentrations were different between the Apex (4.5 mM) and Amplitaq (1.5mM). For background, I originally ran the Apex mastermix with a titration of MgCl2 back in 2010 and had been using a 4.5 mM concentration since then. I decided to test the Amplitaq with the salt concentration recommended in the manufacturer's protocol, which also agreed with a few of the MSAP protocols (such as Liu and Keyte).
- In order to troubleshoot the lack of bands using the Amplitaq mastermix, I increased the salt concentration to 4.5mM MgCl and re-tested the select PCR using sample 97Hpa. I prepared mastermixes using Apex at 4.5mM MgCl2, Apex at 1.5mM MgCl2, and Amplitaq at 4.5mM MgCl2
- Results:

- Increasing the salt concentration in the Amplitaq reaction mixture from 1.5 mM to 4.5 mM MgCl2 resulted in banding similar to that of the Apex mastermix. Of note, the Amplitaq mastermix shows a reduced background. No smearing is observed >1000bp and the bands are sharper in general. The Apex mastermix resulted in banding regardless of the salt concentration, however it does appear that the lower salt concentration does improve the background. I also wanted to investigate the role of annealing temp and cycle number in this PCR. The replicates on the left were run with the TD PCR and a general anneal temp of 56 (25 total cycles of general amplification), the panel on the right the anneal temp was increased to 60C and 15 total cycles. The best resolution on this gel came from the Amplitaq polymerase at 4.5mM MgCl2 and an anneal temp of 56C for 25 cycles
https://www.dropbox.com/s/am7jlckmwqsu9ui/PCR%20080712.jpg
{kind=link}
8/3/12
Summary: Repeat select PCR from 8/2/12 that had contamination in blank reactions. Also include a few reactions using Platinum PCR Taq since I'm not sure if the Apex mmix would be good to put on sequences since it has dye in it.
Procedure:
- prepare mmix using Apex (see MSAP protocol) for 7 reactions
- prepare mmix using Platinum Taq (Invitrogen) for 7 reactions: per rxn: 2.5uL 10x PCR buffer, 0.5uL 10mM dNTP, 0.8uL 50mM MgCl2, 0.5uL each primer, 0.1uL polymerase, 16.1uL H20 (4uL template)
- add template from sample 91 'original 7/26' pre-select PCR (@ 1:20 dilution)
- running the 56C protocol with 25 cycles (same as 8/2/12) and also the 56C protocol with 15 cycles: Each cycling parameters has a replicate of 91H and 91M in both Apex and Invitrogen Platinum and a blank
- here I noticed that the protocol for the '60C' anneal temp that was run yesterday was actually running at 56C. Noted in notebook for 8/2/12
- results:

- Blanks look good, reducing cycle number doesn't really gain anything in terms of high background. The Platinum Taq did not amplify (there is a hint of a few bands in the 91H lane for the 25cycle amplification. I think the issue here may have been the amount of dNTP in the mastermix. I think I need 10mM each, instead I used 10mM total
Amplitaq PCRs
mmix components (per reaction): 2.5uL 10x PCR buffer 2, 2uL 10mM dNTP, 0.5uL each primer (10mM), 0.125uL AmpliTaq polymerase, 0.75uL 50mM MgCl, 13.6uL H20 (25uL reactions, 5uL template).
performing a select PCR (primer pair 3) with Amplitaq - TD PCR w/ 56C anneal
performing pre-select PCR (using sample 97H and 97M) - 1 batch with TD PCR w/ 56C anneal and 1 general preselect protocol with 20cycles at 56C (see MSAP protocol for additional details on parameters).
8/2/12
Summary: Optimizing MSAP PCR reactions to reduce background, using a subset of samples from 7/31/12-8/1/12. Using a touchdown PCR protocol and 2 different annealing temps for the general amplification
Procedure:
- pre-select PCR:
- samples: used digested/ligated DNA from oysters 91 and 152 (7/26/12)
- prepared a single mastermix to run touchdown PCR w/ 2 annealing temps (4uL template, 16uL mmix (mmix prepped per MSAP protocol)
- thermalcycling parameters are below - ran both 91 and 152 samples for the 56C general anneal temp, ran only 91 for the 60C anneal temp
- "56C": 94C 2min, 10 cycles: 94C 30s, 65C 30s (dropping 1C per cycle), 72C 2min, 25 cycles: 94C 30s, 56C* 30s, 72C 2min, 60C 30min
- "60C": varied from above only by increasing anneal temp in 25cycle general amplification (marked by *) to 60C
- results of pre-select: (NOTE 8/3/12: the 60C protocol was in error - both programs ran at 56C)

- diluted all samples by adding 150uL of H20 to the remaining 10uL of PCR product (1:16 dilution)
- compared to initial pre-select PCR run 7/27/12 the smears are more visible, but there is still some banding (1 @ ~1500bp, 1 @ ~900bp)
- select PCR:
- for this test run I am using primer set 3 (wide range of sizes, lots of bands to try to resolve) - (primers: Eco_ACA, Hpa/Msp_TGA)
- prepared a single mastermix to run various pre-select PCR products (27 reactions total w/ 4uL template, 16uL mmix)
- PCR cycling parameters are the same as above, various templates were used:
- "56C" cycling parameters (12 total samples): original pre-select products 7/27/12, original pre-select products with an additional dilution (1:20 total) similar to Liu protocol, pre-select products performed today using 56C protocol (1:16 dilution)
- "60C" cycling parameters (10 total samples): same as 56C for the original pre-select products, but ran pre-select products from 60C protocol (1:16 dilution).
- results of select (NOTE 8/3/12: the 60C protocol was in error - both programs ran at 56C)

- Blanks had contamination. Difficult to say if background has been reduced. Will repeat tomorrow.
8/1/12
Summary: 1) Test run (PCR and gel separation) a few of the unlabeled MSAP primers with the samples prepared earlier this week (EE2 trial) 1) Run PCR products for testing vtg6 and DNMT primers (PCR 7/31/12) on gel.
Procedure:
- selected 2 samples: 91 and 152 to test run 3 (of 7) selective primer pairs
- master mixes for primer pairs 1,2 and 3 were prepared. see MSAP protocol for primer sequences, 40uL reactions were prepared instead of 20uL reactions in order to have enough product to run on 2 gels
- PCR setting as stated in MSAP protocol (Mac>PSTHIRTY thermocycler protocol)
- prepared a 2% metaphor Agarose gel (following mfr instructions) and a 2% Agarose gel
- ran gels for ~1hr at 70V, then ~2 hr at 100V
agarose gel:
primer tests are on the left of the gel, MSAP samples on the right. Primer test samples are (left to right): 3 independent cDNA samples from dg/gonad, gDNA, blank. MSAP samples are (left to right per primer set): 91 Hpa, 91 Msp, 152 Hpa, 152 Msp

metaphorose gel

Conclusions/Next steps:
The vtg primers did not amplify any of the cDNA samples or the gDNA. These primers have been used in Dheilly et al 2012, so I would like to try this PCR again with gonad sample from a late stage female oyster. It's possible the primers are working, but vtg is not expressed in these samples. The DNMT primers amplified the expected size product for the cDNA. The primers do amplify genomic DNA, but it is a larger band indicating these primers span at least 1 intron. The MSAP samples had higher resolution using the metaphorose. There is still a lot of 'smearing' in the lanes. This could possibly be due to background from the first pre-select PCR? In general, the range of band sizes for these primers is quite large between 2000 and 100. It would be difficult to score these bands with the resolution here. Need to find out if these samples are suitable for CE.
7/31/12
Summary: test PCR primers for C.gigas vitellogenin6 and DNMT
Procedure:
- Primer background:
- vtg6 primers (SRID: 1513,1512) based on Dheilly et al 2012
- DNMT primers (SRID: 1511,1510) designed using primer3 based on consensus sequence from 03/9/11
- Prep mastermix for each primer pair (6 reactions): 12.5uL 2xApex, 0.15uL each primer (10uM), 11.2uL H20
- Added 24uL mmix and 1uL template to PCR tube. Templates included:
- Dg/gonad cDNA prepped by Sam 3/11/11 (wells A5, B5, C5)
- gDNA (parent 17 gill sample)
- H20 blank
- Thermocycler parameters: 10min 95C, 40 cycles: 30s 95C, 30s 55C, 60s 72C, 10min 72C
7/27/12
Summary: perform pre-select PCR for MSAP procedure from 7/26/12 (oocyte DNA from EE2 treatment samples)
Procedure:
- end digest-ligation at 8:30am by adding 175uL of PCR grade H20 to reactions
- prepare PCR reactions for pre-select PCR following MSAP protocol (follow link on 7/26 for procedure). Included digest-ligation blanks (digest-ligation reaction with no template) for both HpaII and MspI enzymes as well as PCR blank
- ran 10uL of PCR product on gel and added 90uL of H20 to the remaining 10uL volume

- There was no amplification in the digest-ligation reactions for either enzyme or in the PCR blanks. I am surprised to see the presence of discreet bands in these pre-select samples. Previously, I have only observed the expected smears in the 1200 - 200bp range (e.g. 2/10/11) not discreet bands (but, I have only run pre-select products on the gel twice). I do not think these bands will negatively affect the next round of PCR. Even if these bands 'swamp out' the signal, it will not affect all primer pairs since each combination will amplify different fragments. There is the expected DNA smear observed from about 800-300bp. Although the amount of DNA was normalized in each reaction, there appears to be less PCR product in sample 152. I don't think this will affect subsequent analysis since there was observable amplification and the HpaII sample will only be direction compared to the MspI sample for 152 after selective PCR amplification.
- Next steps: order labeled primers for select PCR in order to run samples on ABI 3730.
7/26/12
Summary: Initiate MSAP for C.gigas oocyte DNA from EE2 treatment samples
Procedure:
- prepare digestion/ligation reactions for the following samples: 91,97,108 (EE2 treated, stage 3), 152 (control stage 2), 153 (control stage 3) following the MSAP protocol
- below are DNA and H20 volumes for each sample in uL

- samples in 37C H20 bath overnight
7/25/12
Summary: complete DNA isolation from 7/24/12
Procedure:
- completed DNA isolation according to DNAzol protocol, eluted samples in H20
- performed EtOH on a subset of samples with low yield (marked w/ asterisk in table below)
- the table below describes (by column): oyster ID, weight of starting tissue, concentration of gDNA, total volume, total yield in ug, required volume for 0.8ug DNA (this is the amount required for MSAP). Only the samples in bold will be used for MSAP

7/24/12
Summary: initiate DNA isolation for 9 C.gigas gamete samples from EE2 trial, 35 day time-point
Procedure:
- sample selection: Nine stripped oocyte samples total. Three 500ng/L EE2 treatment (stage 3), 3 controls (stage 3), 3 controls (stage 2). Rationale: compare treatment v control and also have ability to compare gonad dev. stages 3 v 2.
- EE2 (stage 3): 91 (R373), 97 (R554), 108 (R874)
- control (stage 3): 152 (R373), 157 (R554), 155 (R869)
- control (stage 2): 153 (R373), 159 (R554), 156 (R869)
- Thawed 1 of 2 gamete samples stored at -80C
- Weighed sample, added 500uL DNAzol and 50 pK to all samples with the exception of 153 where 1mL of DNAzol and 100pK was used.
- 91: 18.8ug
- 97: 13.3ug
- 108: 16.8ug
- 152: 16.8ug
- 153: 32.0ug
- 155: 17.9ug
- 156: 9.2ug
- 157: 9.2ug
- 159: ~12ug
- rotating tubes overnight at RT
7/23/12
Summary: staging of gonadal development of 5 week time-point females from control and 500ng/L EE2
Staged 19 female oysters (11 EE2 @ 500ng/L, 8 control) using the staging of Steele and Mulcahy 1999. All but 1 of the oysters were either stage 2 or 3 (a single control oyster was a stage 1). A number of the stage 3 oysters I would like to note with an asterisk because there was some connective tissue remaining within the gonad tissue and some of the follicles had not coalesced. Nevertheless, in terms of this staging system, I would still give them a 3.
| treatment |
individual |
stage |
| 500ng/L EE2 |
91 |
3 |
| 500ng/L EE2 |
96 |
2 |
| 500ng/L EE2 |
97 |
3* |
| 500ng/L EE2 |
98 |
3* |
| 500ng/L EE2 |
108 |
3 |
| 500ng/L EE2 |
110 |
3* |
| 500ng/L EE2 |
112 |
3* |
| 500ng/L EE2 |
115 |
3 |
| 500ng/L EE2 |
116 |
3 |
| 500ng/L EE2 |
119 |
3* |
| 500ng/L EE2 |
120 |
3* |
| control |
152 |
3 |
| control |
153 |
2 |
| control |
155 |
3* |
| control |
156 |
2 |
| control |
157 |
3 |
| control |
159 |
2 |
| control |
166 |
3 |
| control |
172 |
1 |
Statistics: Chi-square between treatment =0.2

took a few more oocyte measurements (see initial measurements 7/20/12) and confirmed that variability is huge. Oysters 152 and 166 are control oysters, all of the others are EE2 treated.

7/20/12
Summary: measurements of oocytes size in EE2 treated v. control oysters
- took 6 images at 400x magnification for each oyster (so far have done this for 13 of 19 oysters) to use for oocyte area analysis
- also have a few 100x images for staging purposes
- at 400x magnification, I can get about 5 oocyte measurements/image. I am only using free oocytes (not attached to the follicular wall) with an observable nucleus (to try to ensure oocytes are on the same plane)
- just to get a feel for the sizes I analyzed the area of 10 total oocytes between 2 images (5/image) for 5 different stage 3 oysters (3 treatment, 2 control). I used ImageJ for this analysis.
- A graph of the sizes are below. The pvalue was 0.3 between treatment and control.
- Initial observations: there is a lot of variability between individuals within treatment. This could at least partly be due to the fact that I only 2 of 6 images were used for the analysis. I observed while taking the images that different portions of the gonad seemed to differ in oocyte size and number - this was making staging more difficult too - so maybe I would get different values when I use all 6 images? This initial look at oocyte area leads me to believe that the variability in this measurement is going to be too high, especially considering the small numbers I have. One question remaining is that at least qualitatively, there appears to be some skew towards a higher stage of gonad development in the treated oysters - would it then be acceptable to pool these stages to compare oocyte area? All of the animals in this initial analysis I would characterize as being stage 3 females.
 l
l7/17/12
Summary: first attempts at measuring oocyte area using NIS elements and ImageJ
NIS Elements
- captured images of oyster 91 and 166 on the microscope in the Young lab at 200x magnification
- manually outlined the area of 10 oocytes manually using the freehand tool in NIS elements - the manual outlining felt a little biased, because the shapes were not round, the perimeter could be biased based on how many points I select as I went around the cell. I would like to try to automate this process.
- areas for the 10 measurements for oyster 91 and 166 are below (note that measurement 7 for oyster 166 is inaccurate)

ImageJ
- used captured images of oyster 91 and 166 on the microscope in the Young lab at 200x magnification
- opened images in ImageJ and made binary image of oyster 166 to facilitate automated area measurements
- used the wand tool to outline regions of interest (ROIs), I wanted to only used oocytes w/ visible nuclei so I had the original image opened side by side
- sometimes the wand tool does not pick up all the edges of the cell, so I had to go in and manually adjust the outline. To do this I select the freehand tool then hold down the option key while circling the area that I do NOT want included.
- it took about 10 minutes to get 15 ROIs highlighted, then I selected all of the RIOs and clicked on measure to get the measurements in pixels
- when I opened the image for oyster 91 it was too difficult to use the wand tool on the binary images because the contrast wasn't strong enough to pick up the cell images.
- Initially, I wanted to use 4 random images of each gonad at 200x magnification and measure ~20 oocytes per image - but because it's difficult to see the outlines, at least on oyster 91, I would like to try using a higher magnification (400 or 600x), to see if the edges are more defined.
- Here are examples of the original and binary images of oyster 91 and 166

7/12/12
Summary: measurements of oocytes using the Young Lab microscope and NIS elements software
- Measured the diameter of the oocytes. since the cells are not round, measurements are from the longest distance of the ovoid shaped oocytes. Averaged ~ 10 measurements per slides, so these measurements are only an estimate and were meant to get familiarized with the software.
- R861 oocytes 30-50um
- R554 oogonium <12um
- R818 early oocytes <15um
- R819 oocytes 40 - 60um
- R371 oogonium <10um
- R385 oocytes 40 - 55um
7/10/12
Summary: Images of day0 histology for staging.
- album of images here: http://www.flickr.com/photos/32788337@N08/sets/72157630520454332/
- slideshow and staging using Steel & Mulcahy 1999 here. I am not confident about R819 and R554 females. I think the shape of the follicles and presence of lots of hemocytes is throwing me off - will try to get another opinion. Follow up: Consensus opinion is that R819 is stage 2 and R554 is stage 1.
Images histology day0
View more PowerPoint from mgavery
7/9/12
Summary: A) Results of actual EE2 concentration in seawater on select days by GCMS; B) Day zero histology notes
Results:
(A) EE2 concentrations in tanks
500ng/L nominal Tank 1:
5/28 (fresh preparation): 350ng/L
5/30 (post-1/2 tank change): 45ng/L
6/4 (24hr post fresh dosing): 25ng/L
50ng/L nominal Tank 3:
5/28 (fresh preparation): 37.2ng/L
5/30 (post-1/2 tank change): 37.4ng/L
6/4 (24hr post fresh dosing): ~37ng/L
(B) Gonad staging of 10 oysters (1 per family) at Day zero of the trial:
NOTE: these are initial notes, looked at slides w/ C. Friedman today, but I need to decide on staging criteria and reexamine
0 - undifferentiated, 1 - very early development, 2-mid development, 3 - ripe, 4-ripe/partial spawn, 5 - reabsorption
R385 - female, stage 2-3 (mature eggs were present in folicle)
R874 - male, stage 3-4 (mature spermatazoa)
R371 - female, stage 1 (very few oocytes in lumen)
R861 - female, stage 2-3
R819 - female 3-4 (ripe)
R373 - male, stage 3-4 (mature spermatazoa)
R869 - male, stage 1-2
R554 - female, stage 5 (possibly spent, in reabsorption, lots of hemocytes)
R363 - male, stage 3-4 (mature spermatazoa)
R818 - female, stage ~2 (no oocytes in middle of follicle, but developing at edge of follicle)
7/6/12
Summary: Quick graphs of sex ratios for day 35 and 42 samples by treatment and by family. Sample sizes vary for all graphs so see raw data/previous notebook entries. Although not significantly different, there is a trend toward more females in treated groups compared to control. Family R554 is the only family that shows a majority of females (for Day 35 & 42 combined: 3/5 (females/total) for control, 3/5 for 50ng/L treatment, and 6/6 for 500ng/L treatment.)


6/21/12
Summary: Documentation of EE2 trial week 6:
The oysters remaining after the 35 day sampling will be maintained for 1 to 2 weeks in the absence of any treatment to assess whether any molecular changes (e.g. DNA methylation), if they occurred, are maintained after the EE2 treatment has been removed.
- 6/21/12 - oysters from replicate tanks were combined into a single tank (all bags from 1 of the tanks were marked with an additional white tag to denote which tank they came from: tanks 2, 4 and 5 were tagged); a complete water change was performed and oysters were fed 4mL.
- 6/22/12 - fed 4mL am and 4mL pm
- 6/23/12 - 1/2 tank water change; fed 6mL
- 6/24/12 - fed 6mL
- 6/25/12 - fed 4mL am and 4mL pm
- 6/26/12 - sampled remaining oysters (52) and determined sex on light microscope. Samples: gill, histology cross-section, gonad
6/14/12 - 6/20/12
Summary: Documentation of EE2 trial week 5:
- 6/14/12 - feed 4mL am and 4mL pm; no mortalities
- 6/15/12 - feed 2mL am and 4 mL pm; full tank water change; mortalities: 1 red tag, Tank 3 (50ng/L) - female
- 6/16/12 - feed 4mL am and 4mL pm; no mortalities
- 6/17/12 - feed 4mL am and 4mL pm; 1/2 tank water change; no mortalities
- 6/18/12 - feed 4mL am and 4mL pm; no mortalities
- 6/19/12 - feed 4mL am and 4mL pm; 1/2 tank water change; no mortalities
- 6/20/12 - Day 35 of trial. Sampled 30 oysters/treatment (15/tank); mortalities: 3 red tag Tank 5 (control), 1 red-blue tag Tank 5 (control)
- oysters were weighed and measured. all data can be found here
- the following samples were taken: gill, gonad, cross-section for histology, gametes
- gamete sampling procedure: animals were stripped of gametes from the gonad tissue (using 0.2µm filtered seawater) on the underside of the animal (this keeps most of the tissue and the gonad on the 'top' of the oyster intact). From this ~5mL sample, the oysters were sexed and two 1mL sub-aliquots were taken after larger particles had settled out. Eggs were centrifuged at 800g for 15 min and spermatazoa were centrifuged at 2500g for 20 min. After centrifugation the seawater was drawn off and the samples were snap frozen.
6/7/12 - 6/13/12
Summary: Documentation of EE2 trial week 4:
- 6/7/12 - feed 4mL am and 4mL pm; 1/2 tank water change; mortalities: 1 not tagged, Tank 1 (500ng/mL) gonad screened and mature spermatazoa obvserved, 1 yellow-green tag Tank 5 control gonad screened and mature spermatazoa observed (in both cases activated sperm were observed after the addition of a drop of seawater to gonad smear on slide)
- 6/8/12- feed 4mL am and 4mL pm; no mortalities
- 6/9/12 - full water change then feed 4mL; no mortalities
- 6/10/12 - feed 4mL am and 4mL pm; no mortalities
- 6/11/12 - feed 4 mL am and 4mL pm; 1/2 tank water change; mortalities: 1 yellow-green tag Tank 3 (50ng/L) - spermatazoa observed, but very little activity.
- 6/12/12 - feed 4mL am and 4mL pm; mortalities: none
- Attempt to thermal spawn: using 1 individual/tank (6 animals total). No oysters spawned, will try again tomorrow. Briefly, was trying to thermal spawn without food. Oysters were placed at 26C and slowly warmed to 30C. Once they were had been at that temp for an hour I added food to see if that would initiate spawning. After another 30 minutes they were all placed back at roomtemp with air to try again tomorrow. If they don't spawn tomorrow I will sample them and look at gametes under scope. Here are some notes (the first 3 digits are the time):

- 6/13/12 - feed 3mL am and 4mL pm; 1/2 tank water change; mortalities: 1 green tag Tank 3 (50ng/L) - female (eggs observed)
- Attempt 2 at thermal spawning (multi-media observations also available):
- 11:30am oysters (in either 200 or 500ml of water) were placed at 28C, over the next 20 min they were brought up to 30C
- 12:30pm no spawning has occurred, sacrificed the Tank 5 oyster to look at gamete maturation. I did a strip spawn of this animal using the 'underside' of the gonad using filtered seawater to rinse gametes into a 50mL conical. I collected about 5mL of gametes. I let the sample settle for a few minutes (there was some heavier material that settled to the bottom), then pipetted 1mL samples (2 total) off the surface of the sample. I put a sample on a slide and was happy to see a really pure egg sample. I spun the 1mL samples at 100g for 15 min, removed the supe and added 1mL 70% EtOH. I looked at the supe and it was cell-free, and the resuspended eggs were still pretty much intact. Since the oysters weren't spawning very easily, I think this is a great substitute to get 'gamete only' samples. The other nice thing is that the oyster remains attached to the cup shell, so that it stays pretty much intact and a nice histology sample can still be taken this way.
- 1:15pm remaining oysters (in their original containers) were cooled slowly in a 20C water bath
- 1:45pm oysters were placed back in heated water bath (water in containers was replaced with 25C water before placed back in 28C waterbath)
- 2:45 observed that one individual, Tank 2, had spawned. Collected 1mL sample from the 500mL of water, this individual was male. Spinning the spermatazoa sample at 100g for 15 min did not work that well, lots of spermatazoa in the supe. I did a second spin at 1000g for 15 min and this sample was 'cell-free'
- The remaining individuals were strip spawned. The quality of the samples varied depending on the size of the oyster, here I also tried strip spawning into weigh boats instead of conical tubes - even though I only have 1 example of sampling into the tube this type of sample seemed more 'pure', I think this is because some of the debris was allowed to settle out before taking the sample. This is how I would like to sample gametes next week.
- Summary of sex of individuals:
- T5 (control) - female
- T6 (control) - male
- T1 (500ng/L) - male
- T2 (500ng/L) - male
- T3 (50ng/L) - female
- T4 (50ng/L) - male
- In summary: Thermal spawning was not that successful, many of the animals had mature gametes (active spermatazoa observed in strip spawned individuals), but still only 1 out of 6 individuals spawned and this was after hours of heating and cooling. This approach will not be viable for obtaining gamete samples from oysters. However, I was pleased with the overall quality of gamete samples that could be obtained by strip spawning. I think the best way to do this is to strip spawn into a 50mL conical tube, let debris settle for a few min, the pipette 1 or 2 (probably 2 to do both RNA and DNA extractions) 1mL samples off the top. To remove the seawater from cells the samples should be spun at 100g for 15 min (may consider doing a higher speed for male samples, but even with low speed a small pellet formed so the quantity of cells should probably be sufficient for DNA extraction). Of the 7 animals assessed today 3 were female and 4 were male. No pattern was observed that indicated a treatment effect.
- Attempt 2 at thermal spawning (multi-media observations also available):
5/31/12 - 6/6/12
Summary: Documentation of EE2 trial week 3: 5/31/12- 6/6/12, cumulative mortality: 4
- 5/31/12 - feed 3.5mL am, 3mL pm, no mortality
- 6/1/12 - feed 3mL am, 3mL pm; 1/2 tank water change; no mortality
- 6/2/12 - feed 3mL am, 3mL pm; no mortality
- 6/3/12 - feed 4mL am; complete water change (fresh working stocks), water samples from Tanks 1 and 3; mortality: 1 red-blue tag Tank 3.
- 6/4/12 - feed 4mL am, 4mL pm; water samples from Tanks 1 and 3; no mortalities
- 6/5/12 - feed 4mL am, feed 4 mL pm; 1/2 tank water change; no mortalities
- 6/6/12- feed 4mL am and pm; mortalities: 1 yellow tag Tank 3 (50ng/L) - no mature gametes observed
5/24/12 - 5/30/12
Summary: Documentation of EE2 trial week 2: 5/24/12 -5/30/12 , cumulative mortality over this period=25
- 5/24/12 - brought in 275 gallons of water yesterday. The room temp had been fluctuating +/- 2 degree for the past few days and the water has seemed to stabilize it a bit. At the start of the trial the temp was set to 18C and there were 11 mortalities in the first week (doesn't appear to be treatment related). The temp was reduced to 16C to attempt to reduce the mortality, but still encourage gametogenesis. I think temp excursions of +/-2 degrees at 16C is more tolerable to the animals. Feeding: 2mL am, 3mL pm Mortalities: 1 red tag Tank 1(500ng/L EE2), 1 unbagged Tank 6 (control), 1 neon yellow tag Tank 5 (control), 1 red-blue tag Tank1 (500ng/L)*, 1 neon yellow Tank 2 (500ng/L)*, 1 orange tag Tank 2 (500ng/L)*, 1 red-blue tag Tank 5 (control)*. Histology was taken for the animals marked w/ and asterisk. red-blue tag Tank 1 had mature eggs under light microscope, red-blue Tank 5 was probably male. Photographs are here.
- 5/25/12 - feed: 3mL am, 3.5mL pm mortalities: 1 green tag Tank 1. Looked at gonad sample under microscope w/ Brent V., no mature gametes present.
- 5/26/12 - feed 3ml am, 3ml pm. mortalities: 1 red-white (red) tag, Tank 1 (500ng/L), 2 red-blue tag Tank 4 (50ng/L), 1 pink tag Tank 4 (50ng/L), changed 1/2 water
- 5/27/12 - feed 3mL am, 3mL pmmortalities: 1 green tag Tank 1 (500ng/L)*, 1 green tag Tank 4 (50ng/L), 1 yellow-green tag Tank 4 (50ng/L)* *=histo
- 5/28/12- feed 3mL am, 3mL pm, complete water change, water samples from Tank 1,3,5 mortalities (all sampled for histo): 1 green tag Tank2 (500ng/L), 1 red-blue tag Tank 2 (500ng/L), 1 yellow tag Tank 4 (50ng/L), 1 pink tag Tank 5 (control)
- 5/29/12-feed 3mL am, water sampled tank 1, 3, 5.
- 5/30/12 - feed 4mL pm (did not feed am because of sampling). Sampled 30 animal/treatment (15/tank) and sampled gill, gonad and histology cross-section. Labels and measurements here. mortalities: green tag Tank 4 (50ng/L), neon yellow Tank 3 (50ng/L), green-yellow tag Tank 2 (500ng/L), blue-red tag Tank3 (50nL/L), green-yellow tag Tank 3 (50ng/L), dark blue tag Tank 4 (50ng/L); changed 1/2 water and took water samples post-change from Tank 1 and 3
5/17/12 - 5/23/12
Summary: Documentation of EE2 trial week 1: 5/17/12 - 5/23/12, temp change from 18-19C, cumulative mortality over this period =11, no more thank 1 animal/tank/day
- 5/17/12 - 4mL algae/tank
- 5/18/12 - temp to 19C, 1/2 tank water changed, 5.5 mL algae/tank (2.5mL am, 3 mL pm), mortalities: 1 unbagged Tank 4 (50ng/mL EE2)
- 5/19/12 - 4 mL algae/tank pm
- 5/20/12 - 1/2 tank water changed, 4mL algae/tank pm
- 5/21/12 - 4mL feed (2mL am, 2mL pm), mortalities: 1 red-blue tag, Tank 6 (control); 1 orange tag, Tank 5 (control), 1 red tag, Tank 2 (500ng/L EE2), 1 yellow-green tag, Tank 1 (500ng/L)
- 5/22/12 - 5mL feed (2mL am, 3 mL pm), full water change, mortalities: 1 red tag, Tank 4 (50ng/L); 1 pink tag, Tank 3 (50ng/L); 1 yellow tag, Tank 2 (500ng/L); 50mL water samples from tanks 1 and 3 frozen at -20C for EE2 analysis
- 5/23/12 - 5mL feed (2mL am, 3mL pm), mortalities: 1 red tag, Tank 5 (control); 1 light blue tag, Tank 4 (50ng/L), Tank 3 neon yellow (50ng/L). It looked like they had cleared the 2mL from the morning so were fed 3 mL in the afternoon; 50mL water samples from tanks 1 and 3 frozen at -20 for EE2 analysis.
5/16/12
Summary: Day 0 of EE2 trial.
Procedure:
- On 5/10/12, 300 oysters were brought into the lab from Thorndyke Bay. They are Pipestem (BC) oysters, 30 each from 10 different single pair matings. Information on the individual families can be found here.
- Oysters were placed in a single container at 16C and fed daily. After 4 days (5/14/12) the temperature was raised to 18C.
- The experiment was initiated on 5/16/12 and included 1) day zero sampling, 2) preparation of EE2 stock solutions and 3) distribution of oysters into tanks.
- Day zero sampling:
- 1 oyster from each family, n=10
- oysters were measured, weighed and then shucked (see below for measurements)
- for sampling the whole body was removed gentry from the shell and a cross-section of the oyster, half-way between the muscle and the labial palps was taken for histology
- the following tissues were then sampled for DNA/RNA analysis: digestive gland, mantle, muscle, gill and stored immediately at -80C
- photographs were taken of each oyster (with the exception of R385). Three of the animals appeared to have initiated gametogenesis (white fluid was released after cross-sectioning).
- Preparation of EE2 stock solutions:
- prepared a 10mg/mL EE2 stock solution: 0.0601g in 6mL EtOH
- prepared a 1mg/mL EE2 stock solution: 0.0051g in 5mL EtOH
- stock solutions were mixed for 1 hr on a stir plate, these stock solutions should be stable >1yr.
- prepared 200x dilutions of each stock in DI water to make the working stock solutions (as well as an EtOH control): 10uL each stock or EtOH only, 4,990uL DI H20. *Working stocks will be made fresh every week
- For the 500ng/L treatment tanks, 750uL of the 0.02mg/mL working stock was added to a final volume of 30L (15,000 total ng or 500ng/L) *additions were made by adding 750uL to the final 5L of water and swirling to mix before adding to the tanks.
- For the 50ng/L treatment tanks, 750uL of the 0.002mg/mL working stock was added to a final volume of 30L (1,500 total ng or 50ng/L)
- For the control tanks, 750uL of the EtOH only working stock was added to the tanks
- original worksheets can be found here
- Distribution of oysters into tanks:
- oysters from individual families are being kept separate in small mesh bags for this experiment. for each family:
- 9 oysters from each family were placed into the 500ng/L treatment (2 replicate tanks, so 5 animals into 1 tank and 4 into the replicate tank), 9 oysters will be placed into the 50ng/L treatment (same distribution between tanks). The families are separated in small mesh bags and so are distributed by family for a total of 45 oysters per tanks (see layout -page 2)
- 8 oysters from each family were distributed between the 2 replicate control tanks
- 5 additional oysters (not separated by family) were placed into the control tank to equal 45 oysters/tanks. These additional oysters may be opened periodically to check for ripeness
- additional oysters 16 total were distributed among the tanks (not separated by family) and may also be used to check for ripeness
- families can be identified by color coding of zipties on bags, additionally a small tube was added to each bag with the family number on it.
- animals were fed (1.5mL/container)
- oysters from individual families are being kept separate in small mesh bags for this experiment. for each family:
Day zero measurements:
length and width in mm, mass in grams. original data sheet here.

Oyster bag labeling scheme:

4/10/12
Summary: summarize of results for most recent Nanostring samples 3/21/12.
This table is tracking the results of a number of the more interesting (i.e. there is evidence of methylation and/or differential methylation) sites. The most recent samples, tested 3/21/12, include additional gonad samples and larvae samples as we were interested in the differential methylation observed for the hexokinase loci (probe ID: EU342886_1129).

3/1/12
Summary: results of bisulfite sequencing for C.gigas hexokinase gene fragment for: larvae, sperm, gill and EE2 exposed gill
NOTES: see original notes from sequencing 2/21/12. The sequencing facility was contacted and had some thoughts about why the sequencing results were poor (see email here) and they re-ran the samples. A summary of the results are below:
Percent methylation is based on the results of 4 individual clones analyzed for each sample (between 2 and 6 sequencing replicates were analyzed for each clone). For the samples noted w/ an asterisk only 2 of the 4 clones were analyzed at that positions (for the other clones this position either had a SNP or deletion so that it was not a CpG site), for the EE2 exposed gill none of the clones could be analyzed at position 2. For that reason I do not have a lot of confidence in the results for position 2. For the larvae samples, a similar situation was observed for positions 3 and 4 so that only 2 of the 4 clones could be analyzed. Position 4 CpG is the site that is being analyzed in Nanostring (EU342886_1129).

In summary, I am confident in saying that this region is differentially methylated between sperm and gill (and trends point that way for larvae too). Other trends observed are that it appears that the this region has higher methylation in the larvae sample compared to the gill, and that the EE2 exposure does not appear to affect methylation in this region of the hexokinase gene - but it is important to note that the gill sample analyzed was not the 'control' for the EE2 experiment, but rather a gill sample taken at a different time. I would like to get additional clones analyzed for all 4 of these samples (especially the larvae) to have more confidence in the quantitation of these results. Comparing the results of CpG position 4 with that of Nanostring results the trends are the same (see entry 12/21/11) although quantitatively different. In summary, Nanostring results for all gill samples tested (including EE2 exposed gill) had 0% methylation, the larvae had 63% methylation and the sperm showed 30% methylation. It may be important to note that if there is a real SNP in this position (as it appears there may be from some of the larvae clones) it's possible that the %methylation result from Nanostring could be biased because the enzyme recognition site may be absent.
2/23/12
Summary: complete gDNA isolation from 2/22/12; send samples to Nanostring
Procedure:
- larvae samples looked pretty good after digestion O/N. Vortexed all samples briefly before centrifuge step. Followed DNAzol procedure including 2x washes in 75% EtOH
- solubilized samples in H20. Female gonad samples had a small pellet so solubilized in 100uL. Male gonad and larvae sampled solubilized in 100uL (larvae had been split in half so 200uL total/sample). There was still some color to the larvae pellet, so following the note in the DNAzol procedure that 'some insoluble material may remain' after solubilization I spun samples at 12k g for 10min and removed the supernatant. All samples were now clear and free of particulates.
- Combined the replicate larvae samples and Quantified all samples on the Nanodrop

- The larvae extractions worked pretty well. I think rinsing the larvae first, having sufficient DNAzol (1mL opposed to 500uL last time) and crushing the larvae w/ a pestle before digestion helped. I also think the centrifugation step at the end to remove the insoluble material helped to clean up the DNA. The concentration of the female gonad samples were a bit low for the Nanostring assay so initiated an EtOH precipitation to concentrate these samples:
- added 0.1 vols of 3M sodium acetate to 280uL of sample, mixed and then added 2.5 vols of 100% EtOH. Mixed and incubated @ -80C for 4hr. Pelleted DNA 16,000g, 15mins, 4C. Discarded supe. Washed pellet w/ 1mL 70% EtOH (2x washes). Pelleted DNA 16,000g, 5mins, 4C. Discarded supe. Resuspended pellets in a total of 30uL H20 and spec'd.
- P28 gonad female - 260.45ng/uL, P30 gonad femaile - 187.0ng/uL
- added 0.1 vols of 3M sodium acetate to 280uL of sample, mixed and then added 2.5 vols of 100% EtOH. Mixed and incubated @ -80C for 4hr. Pelleted DNA 16,000g, 15mins, 4C. Discarded supe. Washed pellet w/ 1mL 70% EtOH (2x washes). Pelleted DNA 16,000g, 5mins, 4C. Discarded supe. Resuspended pellets in a total of 30uL H20 and spec'd.
- Shipped 4 samples to Nanostring:
- larvae 4-19-11 155: 20uL @ 479.8 ng/uL
- larvae 4-19-11 159: 20uL @ 533.62 ng/uL
- P10 gonad male (vinclozolin treated): 15uL @901.9ng/uL
- P28 female: 20uL @260.45 ng/uL
2/22/12
Summary: begin gDNA isolation for C.gigas samples (gonad and larvae) to be tested in the last batch of Nanostring assays.
The goal is to get additional larvae and gonad sample to test using the Nanostring assay since we only currently have data for 1 sample of each. I will be isolating fresh DNA from the gonad samples I have from the July 2010 spawning as well as some larval samples from the hatchery (4-19-11 155 and 4-19-11 159).
Procedure:
- gonad samples: only had 2 sperm samples to choose fromm1 was 5-aza treated the other vinclozolin treated, I selected the vinclozolin treated (P10). For the female samples, I selected 2 control females with the most tissue remaining (P30 and P32). I added half of the remaining sample (<25mg) to a tube and added 1mL DNAzol and 400ug pK to incubate RT/rotating. I also used a pestle to try to break up the sperm sample.
- larvae samples. The larvae were stored in RNAlater so they were rinsed in 2x in 1mL TE (decanting between rinses) to remove the residual RNAlater. Samples were divided in half and crushed them with a pestle in 1mL of DNAzol. 400uL of pK were added and samples were incubated at RT/rotating.
- next step: will complete the isolation after incubating overnight.
2/21/12
Summary: notes from Sanger sequencing results from samples sent 2/2/12
Results: Overall the quality of the sequences were surprisingly low. I spot checked the concentration of the plasmids on the Nanodrop and they are consistent and of sufficient conc (all between 150 - 250ng/uL). There are 3 sequencing reps per clone (2F and 1 R using the M13 primers). For many of the plasmids one or 2 of the replicates had decent sequences but the third is horrible. Indicating the plasmid is ok and either the loading or the sequencing was bad? The hexokinase sequences were mediocre overall. For the Vtg plasmids there was consistent poor quality. There was only good sequence data for the 3' sperm sample (the 5' sperm and 3' and 5' EE2 sample did not have any good quality reps). Summaries for each gene are below:
{kind=link}
Hexokinase: more clones needed but the general trend follows the Nanostring results with both gill samples showing zero methylation at the Nanostring target (3/3 clones) and at least some methylation in the larvae (1/3) and sperm (3/3) at the same site. More clones are needed to see if the trends hold true. Next steps: Submit samples for sequencing again, select final 4 samples for Nanostring analysis. At this point I would like to test a 2nd gonad sample for both sperm and ovary as well a separate C.gigas larvae sample. For the 4th sample it may be interesting to include 1 rep of the gills samples exposed to L-methionine.
Vitellogenin: only the 3' sperm sample had good sequence consensus (shown below). These sequences align w/ what I was considering the 5' region of this nested primer set (labeling problems?). Most of the CpG sites here are unmethylated. *Cytosines are blue, CpGs are marked in purple on CDS - no blue at CpG sites = no methylation* There are a small number of putative 'unconverted' cytosines (C's in non-CpG context and only 1 clone). Next steps: Submit samples for sequencing again. Right now the results are just descriptive so it would be nice to get results to compare to the methylation status of the sperm sample.

2/2/12
Summary: completed mini-preps from 2/1/12
Procedure: repeated procedure from 1/28/12
Next Step: load sequencing plate and send Vtg and hexokinase samples out for Sanger sequencing on 2/6/12
2/1/12
Summary: run gel for PCR screen of vitellogenin bisulfite clones, initiate mini-prep
Results:
0.8% agarose gel, see 1/31/12 for PCR

Most of the clones look good. The expected band size (+vector sequence (used M13 primers)) for vtg 5' is ~700bp, and vtg 3' is ~600bp.
1/31/12
Summary: cloning of C. gigas vitellogenin bisulfite treated DNA cont. from 1/30/12. Today: select colonies and PCR-screen for insert using M13 vector primers.
Procedure: repeated procedure from 1/26/12 (A)
1/30/12
Summary: initiate cloning of bisulfite treated C.gigas DNA for the (putative) last exon of Vitellogenin. Samples: EE2 exposed gill and sperm (see 1/123/12 for PCR)
Procedure: repeated procedure from 1/25/12 here. The PCR products here are from a nested bisulfite primers - see 12/6/11 for sequences
1/28/12
Summary: finish mini-prep of plasmids from 1/27/12 (hexokinase bisulfite treated 4 C.gigas samples). CLC: Started CLC jobs notes for today here
Procedure
Decanted ~1.5mL broth from each tube into microcentrifuge tube and spin max speed 1min. Remove supe. Decant an additional ~1.5 mL broth into tube, spin max 1min. Decant supe. Follow instructions for Qiagen's Qiaprep Spin Miniprep kit. NOTE: did not perform step 10 which is a wash w/ Buffer PB. Stored tubes in labeled rack in large -20C.
Next Steps: Will clone vitellogenin PCR products (see 1/13/12) next week then submit all the samples on a plate for sequencing (probably Thursday).
1/27/12
Summary: run gel for PCR screen of hexokinase bisulfite clones, initiate mini-prep
Results:
0.8% agarose gel, see 1/27/12 for PCR

PCR screen looks good for a majority of the clones. The predicted band size from the hex PCR is 503bp, plus the ~200 bp of vector (used M13 primers) gives a 700bp, which is where a majority of the clones are (using HyperladderII). Note: I was not able to load sperm clone 8 or EE2 1 (human errors) so there is nothing loaded in those lanes. I will pick 4 clones each w/ bands @ 700bp to do mini-preps.
MiniPrep:
added 5mL liquid LB broth + 50ug/mL Kan to individual tubes then used a toothpick to innoculate broth w/ the selected re-streaked colonies. tubes were placed in 37C shaking at 220rpm to incubate over night
1/26/12
Summary: A) cloning of C. gigas hexokinase bisulfite treated DNA cont. from 1/25/12. Today: select colonies and PCR-screen for insert using M13 vector primers. B) notes for de novo assembly of C.gigas fosmid reads.
Procedure:
A)
- there were white and blue colonies for each of the 4 samples. For each sample, 10 while colonies were re-streaked and PCR-screened using M13 primers. Forgot to put X-gal on the plates used for re-streaking again (ok, hopefully that will be the last time now)
- prepared mastermix (50uL reactions) with 25uL 2x Apex mmix, 0.3uL each F and R primer, 24.4uL H20
- PCR parameters
- 95C 10 min
- 40 cycles
- 95C 30 sec
- 55C 30 sec
- 72C 120 sec
- 72C 10 min
General notes: Focus on assembling 1 fosmid at a time first, ignore the paired reads, do not need to trim reads first
1. Started assembly w/ fosmid ..17. I will try to do a few assemblies with ..17 (varying parameters), before going onto other fosmids
2. de novo assembly: fosmid ..17 paired end reads. Mostly default parameters: mismatch=2, insertion=3, deletion=3, length fraction=0.5, similarity=0.8, distance: min-150, max-200 (did not check the 'guidance only' box), 'vote', 'random', minimum length of contig=100
1/25/12
Summary: initiate cloning of C. gigas hexokinase bisulfite treated DNA
Procedure:
- Sample: hexokinase 5' PCR product (see target sequence below) for 4 samples: gill, sperm, EE2 exposed gill, larvae (see 1/13/12 for PCR)
- thaw bands, transfer to ultra-DA purification tubes, spin at 5000rcf for 10 min
- warm plates to RT
- prepare cloning rxn (for each sample): 2.8uL PCR, 0.7uL salt soln, 0.7ul vector
- rxns incubated at RT for 12min, then placed on ice
- add 2uL rxn to vial of TOP10 competent cells
- incubate on ice for 20 min
- heat shocked at 42C for 30 sec, then back on ice quickly
- add 250uL RT SOC medium
- incubate tubes horizontally at 37C for 1 hr, at 200rpm
- in the meantime, added 40uL of 40mg/mL X-gal onto 7 room temp plates then dried at 37C
- after incubation spread cell broth onto 2 plates (150uL and 100uL respectively)
- incubate at 37C overnight
1/13/12
Summary: Second round PCR and band excision for bisulfite sequencing of portions of C. gigas hexokinase and Vtg genes.Continued from 1/12/12
Procedure:
- 2nd round PCR
- 1uL of PCR product from the 1st round PCR
- had 2 blanks for each reaction: 1uL of blank from 1st round (1º), and 1uL H20 (2º)
- master mix: same as above
- 4 primer pairs(SRID in parenthesis)
- Starting w/ 1st round Vtg primers:
- Cg_Vtgbs_lastout_F & Cg_Vtgbs_lastin_R(1435, 1432)
- Cg_Vtgbs_lastin_F & Cg_Vtgbs_lastout_R(1433, 1434)
- Staring w/ 1st round hexokinase primers
- Cg_hexkinBS_out_F & Cg_hexkinBS_inA_R(1443, 1440)
- Cg_hexkinBS_inB_F & Cg_hexkinBS_out_R(1439, 1442)
- Starting w/ 1st round Vtg primers:
- cycling parameters: same as 1st round PCR (see 1/12/12)
all PCR rxns run out on a 0.8% agarose gel
order of samples (left to right) for each primer set: gill E, sperm, gill EE2 exposed, larvae (control), blank 1º, blank 2º

All major bands were excised from the gel and stored @ -20 (Mac's bisulfite box). The major bands were all the expected size. This was the first time these hexokinase primers were tested. The 3' pair of these nested primers showed a faint band @ ~300bp, but the major bands still have pretty good intensity.
Conclusions/Next steps: It appears from the band sizes that the primers are working as expected. The next step is to do some cloning with a sub-set or maybe all? of these samples.
1/12/12
Summary: First round PCR for bisulfite sequencing of portions of C. gigas hexokinase and Vtg genes. The samples: gillE, gill EE2 100ng/L (3), sperm (P19), larvae (control from 5-aza experiment)
Procedure:
- 1st round PCR
- used 1uL of template and 24uL mmix composed of : 12.5uL 2x Apex, 0.3uL each 10uM primer stock and 10.9uL H20 per rxn.
- primers used: Vitellogenin: Cg_Vtgbs_lastout F & R (SRID: 1435, 1434), Hexokinase: Cg_hexkinBS_out F & R (SRID: 1443, 1442)
- 95C 4min
- 5 cycles: 95C 30seconds, 52C 90seconds, 72C 120 seconds
- 25 cycles: 95C 30seconds, 52C 90seconds, 72C 90 seconds
- 72C 4min
- 4C hold
Next Steps: Will do 2nd round PCRs tomorrow.
1/11/12
Summary: Bisulfite conversion of 8 samples C.gigas gDNA (a majority have been previously tested on the nCounter system)
Procedure
- dissolve bisulfite mix (1 tube) w/ RNAse free water (per mfr instructions)
- add DNA and H20 to a final vol of 20uL and NMT 2ug DNA PCR tubes (mfr says 0.2mL tubes, but I didn't have those)
- gill E (samples A - H isolated for Nanostring last year): 16.4uL (@121.58ng/uL), 3.6uL H20
- control larvae (from 5-aza experiment): 15.6uL (@128.02ng/uL), 4.4uL H20
- 5-aza larvae: 5.7uL (@352.9ng/uL), 14.3uL H20
- sperm (P19 gonad control): 18.4uL (@108.5ng/uL), 1.6uL H20
- ovary (P17 vinclozolin tx): 8.7uL (@230.2ng/uL), 11.3uL H20
- gill EE2 (100ng/L, rep 3): 13.0uL (153.62ng/uL), 7.0uL H20
- gill L-MET (l methionine 48h, rep d): 20uL (@ 97.99ng/uL)
- gill low pH (FISH 441 alive diploid low pH): 11.4uL (@ 175.34ng/uL), 8.6uL H20
- added bisulfite mix and DNA protection buffer (manual says DNA protect buffer should be green, it was more brown, the color still turned blue (as expected) once everything mixed)
- performed conversion in thermal cycler and cleaned up DNA per mfr's protocol. Only 1 elution was performed in a total vol of 20uL.
- spec'd samples on Roberts Lab nanodrop to estimate quantity and check quality


I used the wrong constant for gDNA, so the quant is just an estimate. The A260/320's are low for some of the samples and the yields are really variable. Just to point out, the last time I did this procedure the A280/A320 was ~3 (12/10/09) and the yields were more consistent. Next step, I am going to use some of these samples to do bisulfite sequencing of a portion of the hexokinase gene that has been assayed on the nCounter system and showed differential methylation between gill and all other tissue samples. I am going to do the bisulfite PCRs for the Vtg primers at the same time since I have them.